 |
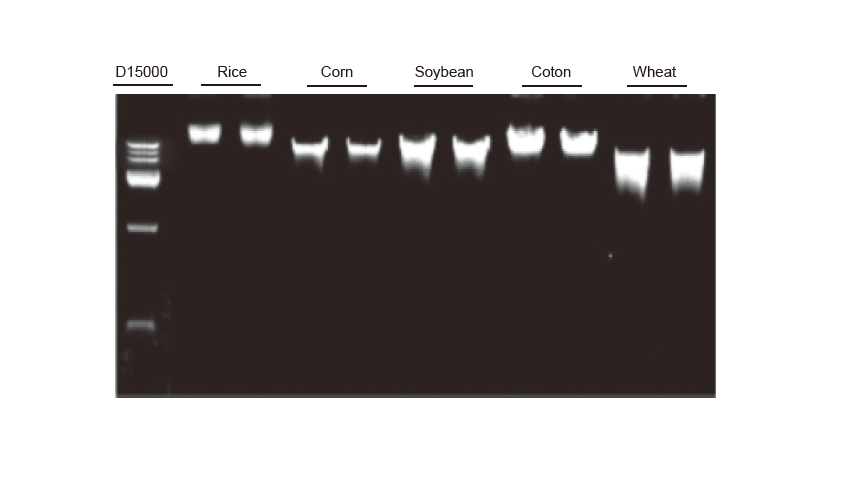
Genomic DNA extraction Genomic DNA extraction was performed on 100 mg leaves of rice, corn, soybean, cotton and wheat, respectively. The experiment was repeated twice. 3 μl DNA from the total 100 μl eluents was loaded per lane. The concentration of the agarose gel was 2%. The electrophoresis was performed under 6 V/cm for 20 min. D15000: TIANGEN D15000 DNA Marker. |
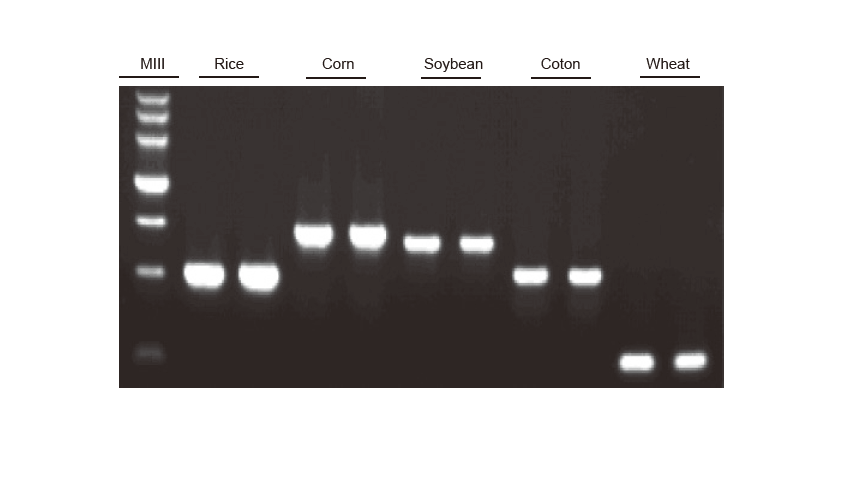 |
PCR Detection Genomic DNA of rice, corn, soybean, cotton and wheat were amplified, respectively. The experiment was repeated twice. 6 μl from the total 20 μl reaction system was loaded per lane. The concentration of the agarose gel was 2%. The electrophoresis was performed under 6 V/cm for 20 min. D15000: TIANGEN D15000 DNA Marker. |
GMO Crop Extraction & Amplification Kit
Features
■ Wide applicability: This kit can extract high quality genomic DNA from five major GMO crops.
■ Simple and fast: GMO crop genomic DNA extraction can be completed within 2 hours. No need for large refrigerated centrifuges, low requirements for instruments and equipment. Suitable for rapid genomic DNA extraction of GMO crops at all levels of research institutions.
■ High efficiency and specificity: The unique buffer of the antibody-modified Taq polymerase ensures efficient polymerase amplification, which is more specific than normal Taq polymerase.
Applications
The kit can extract high quality genomic DNA from major GMO crops such as wheat, corn, rice, cotton and soybean, and perform transgenic PCR detection on GMO crops.
All the products can be customized for ODM/OEM. For details, please click Customized Service(ODM/OEM)
Experimental Example
FAQ
A-1 Template
■ The template contains protein impurities or Taq inhibitors, etc. ——Purify DNA template, remove protein impurities or extract template DNA with purification kits.
■ The denaturation of template is not complete ——Appropriately increase denaturation temperature and prolong denaturation time.
■ Template degradation ——Re-prepare the template.
A-2 Primer
■ Poor quality of primers ——Re-synthesize the primer.
■ Primer degradation ——Aliquot the high concentration primers into small volume for preservation. Avoid multiple freezing and thawing or long-term 4°C cryopreserved.
■ Inproper design of primers (e.g. primer length not sufficient, dimer formed between primers, etc.) -Redesign primers (avoid formation of primer dimer and secondary structure)
A-3 Mg2+concentration
■ Mg2+ concentration is too low ——Properly increase Mg2+ concentration: Optimize the Mg2+ concentration by a series of reactions from 1 mM to 3 mM with an interval of 0.5 mM to determine the optimal Mg2+ concentration for each template and primer.
A-4 Annealing temperature
■ The high annealing temperature affects the binding of primer and template. ——Reduce the annealing temperature and optimize the condition with a gradient of 2°C.
A-5 Extension time
■ Short extension time——Increase extension time.
Phenomena: Negative samples also show the target sequence bands.
A-1 Contamination of PCR
■ Cross contamination of target sequence or amplification products ——Carefully not to pipet the sample containing target sequence in the negative sample or spill them out of the centrifuge tube. The reagents or equipment should be autoclaved to eliminate existing nucleic acids, and the existence of contamination should be determined through negative control experiments.
■ Reagent contamination ——Aliquot the reagents and store at low temperature.
A-2 Primer
■ Mg2+ concentration is too low ——Properly increase Mg2+ concentration: Optimize the Mg2+ concentration by a series of reactions from 1 mM to 3 mM with an interval of 0.5 mM to determine the optimal Mg2+ concentration for each template and primer.
■ Improper primer design, and the target sequence has homology with the non-target sequence. ——Re-design primers.
Phenomena: The PCR amplification bands are inconsistent with the expected size, either large or small, or sometimes both specific amplification bands and non-specific amplification bands occur.
A-1 Primer
■ Poor primer specificity
——Re-design primer.
■ The primer concentration is too high ——Properly increase the denaturation temperature and prolong denaturation time.
A-2 Mg2+ concentration
■ The Mg2+ concentration is too high ——Properly reduce the Mg2+ concentration: Optimize the Mg2+ concentration by a series of reactions from 1 mM to 3 mM with an interval of 0.5 mM to determine the optimal Mg2+ concentration for each template and primer.
A-3 Thermostable polymerase
■ Excessive enzyme amount ——Reduce enzyme amount appropriately at intervals of 0.5 U.
A-4 Annealing temperature
■ The annealing temperature is too low ——Appropriately increase the annealing temperature or adopt the two-stage annealing method
A-5 PCR cycles
■ Too many PCR cycles ——Reduce the number of PCR cycles.
A-1 Primer ——Poor specificity ——Re-design the primer, change the position and length of the primer to enhance its specificity; or perform nested PCR.
A-2 Template DNA
——The template is not pure ——Purify the template or extract DNA with purification kits.
A-3 Mg2+ concentration
——Mg2+ concentration is too high ——Properly reduce Mg2+ concentration: Optimize the Mg2+ concentration by a series of reactions from 1 mM to 3 mM with an interval of 0.5 mM to determine the optimal Mg2+ concentration for each template and primer.
A-4 dNTP
——The concentration of dNTPs are too high ——Reduce the concentration of dNTP appropriately
A-5 Annealing temperature
——Too low annealing temperature ——Appropriately increase the annealing temperature
A-6 Cycles
——Too many cycles ——Optimize the cycle number

The first step is to choose the appropriate polymerase. Regular Taq polymerase cannot proofread due to lack of 3’-5’ exonuclease activity, and mismatch will greatly reduce the extension efficiency of fragments. Therefore, regular Taq polymerase cannot effectively amplify target fragments larger than 5 kb. Taq polymerase with special modification or other high fidelity polymerase should be selected to improve extension efficiency and meet the needs of long fragment amplification. In addition, the amplification of long fragments also requires corresponding adjustment of primer design, denaturation time, extension time, buffer pH, etc. Usually, primers with 18-24 bp can lead to better yield. In order to prevent template damage, the denaturation time at 94°C should be reduced to 30 sec or less per cycle, and the time to rise temperature to 94°C before amplification should be less than 1 min. Moreover, setting the extension temperature at about 68°C and designing the extension time according to the rate of 1 kb/min can ensure effective amplification of long fragments.
The error rate of PCR amplification can be reduced by using various DNA polymerases with high fidelity. Among all the Taq DNA polymerases found so far, Pfu enzyme has the lowest error rate and the highest fidelity (see attached table). In addition to enzyme selection, researchers can further reduce PCR mutation rate by optimizing reaction conditions, including optimizing buffer composition, concentration of thermostable polymerase and optimizing PCR cycle number.




× ![]()
Write your message here and send it to us
Products categories
WHY CHOOSE US
Since its establishment, our factory has been developing first world class products with adhering the principle
of quality first. Our products have gained excellent reputation in the industry and valuabletrusty among new and old customers..
- Tel: +86 010-59822688
- Building 5, No. 86, Shuangying West Road, Changping District, Beijing.
- people@tiangen.com




© Copyright - 2010-2021 : All Rights Reserved. Hot Tags, Hot Products, Sitemap
Ctdna Extraction Kit, Circulating Dna Extraction Kit, Viral Dna Extraction Kit, Dna Extraction Kit, Cell Free Dna Extraction Kit, Gdna Extraction Kit,
Ctdna Extraction Kit, Circulating Dna Extraction Kit, Viral Dna Extraction Kit, Dna Extraction Kit, Cell Free Dna Extraction Kit, Gdna Extraction Kit,