At present, high-throughput sequencing technology is mainly based on next generation sequencing technology. As the reading length of next generation sequencing technology is limited, we must break the full length sequence into small fragment libraries to sequence. According to the needs of different sequencing experiments, we usually choose single-ended sequencing or double-ended sequencing. Currently the DNA fragments of the next generation sequencing library are generally distributed in the range of 200-800 bp.
a) DNA is poor in quality and contains inhibitors. Use high-quality DNA samples to avoid inhibition of enzyme activity.
b) The amount of DNA sample is insufficient when using PCR-free method to construct DNA library. When the input of the fragmented DNA exceeds 50 ng, PCR-free workflow can be selectively carried out during the library construction process. If the copy number of the library is too low to be directly sequenced, the DNA library can be amplified by PCR after the adapter ligation.
c) RNA contamination leads to inaccurate initial DNA quantification RNA contamination may exist in the purification process of genomic DNA, which may lead to inaccurate DNA quantification and insufficient DNA loading during library construction. RNA can removed by treating with RNase.
A-1
a) Small fragments (60 bp-120 bp) appears Small fragments are usually adapter fragments or dimers formed by adapters. Purification with Agencourt AMPure XP magnetic beads can effectively remove these adapter fragments and ensure sequencing quality.
b) Large fragments appear in the library after PCR amplification The size of the library DNA fragment will increase by 120 bp after the adapter is ligated. If the DNA fragment increases by more than 120 bp after the adapter ligation, it might be caused by abnormal fragment amplification of excessive PCR amplification. Reducing the number of PCR cycles can prevent the situation.
c) Abnormal size of library DNA fragments after adapter ligation The length of the adapter in this kit is 60 bp. When the two ends of the fragment are ligated to the adapters, the length will only increase by 120 bp. When use an adapter other than that provided by this kit, please contact the supplier to provide relevant information such as adapter length. Please ensure that the experiment workflow and operation follow the steps described in the manual.
d) Abnormal DNA fragment size before the adapter ligation The reason for this problem may be caused by wrong reaction conditions during DNA fragmentation. Different reaction times should be used for different DNA input. If the DNA input is more than 10 ng, we recommend to choose the reaction time of 12 min as the starting time for optimization, and the fragment size produced at this time is mainly in the range of 300-500 bp. Users can increase or decrease the length of DNA fragments for 2-4 min according to their own requirements to optimize the DNA fragments with the required size.
A-2
a) Fragmentation time is not optimized If the fragmented DNA is too small or too large, please refer to the Guidelines for Fragmentation Time Selection provided in the instruction to determine the reaction time, and use this time point as a control, additionally set up a reaction system to prolong or shorten 3 min to make more accurate adjustment on fragmentation time.
A-3
Abnormal size distribution of DNA after fragmentation treatment
a) Incorrect thawing method of fragmentation reagent, or the reagent is not completely mixed after thawing. Thaw the 5× Fragmentation Enzyme Mix reagent on ice. Once thawed, mix the reagent evenly by gently flicking the bottom of the tube. Do not vortex the reagent!
b) The DNA input sample contains EDTA or other pollutants Depletion of salt ions and chelating agents in the DNA purification step is particularly important for the success of the experiment. If DNA is dissolved in 1×TE, use the method provided in the instruction to perform fragmentation. If the EDTA concentration in the solution is uncertain, it is recommended to purify the DNA and dissolve it in deionized water for subsequent reaction.
c) Inaccurate initial DNA quantification The size of fragmented DNA is closely related to the amount of DNA input. Before fragmentation treatment, accurate quantification of DNA using Qubit, Picogreen and other methods is essential to determine the exact amount of DNA in the reaction system.
d) The preparation of the reaction system does not follow the instruction The preparation of fragmented reaction system must be carried out on ice strictly according to the instructions. To ensure the best effect, all reaction components should be placed on ice and the preparation of reaction system should be carried out after complete cooling. After the preparation is completed, please flick or pipet to mix thoroughly. Do not vortex!
1. Improper mixing method (vortex, violent oscillation, etc.) will cause abnormal distribution of library fragments (as shown in the following figure), thus affecting the quality of the library. Therefore, when preparing the Fragmentation Mix reaction solution, please gently pipette up and down to mix, or use the fingertip to flick and mix evenly. Be careful not to mix with vortex.
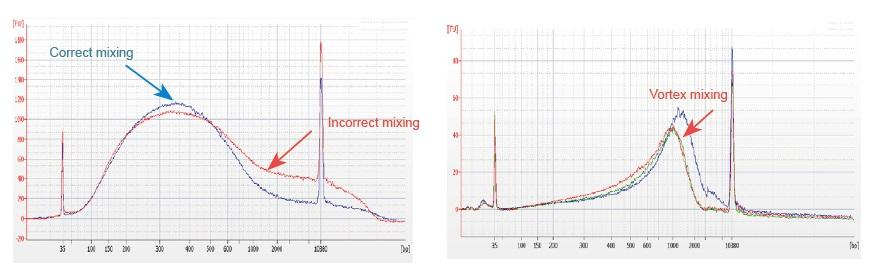
2. High purity DNA must be used for library construction
■ Good DNA integrity: The electrophoresis band is more than 30 kb, without tailing
■ OD260/230: >1.5
■ OD260/280: 1.7-1.9
3. DNA input amount must be accurate It is suggested to use Qubit and PicoGreen methods to quantify DNA, rather than Nanodrop.
4. The content of EDTA in DNA solution must be determined EDTA has a great influence on the fragmentation reaction. If the content of EDTA is high, DNA purification needs to be performed before the subsequent test.
5. The fragmentation reaction solution must be prepared on ice The fragmentation process is sensitive to reaction temperature and time (especially after adding enhancer). In order to ensure the accuracy of reaction time, please prepare reaction system on ice.
6. The fragmentation reaction time must be accurate The reaction time of the fragmentation step will directly affect the size of the fragment products, thus affecting the size distribution of DNA fragments in the library.
1. What type of sample is applicable to this kit?
The applicable sample type of this kit can be total RNA or purified mRNA with good RNA integrity. If total RNA is used to construct the library, it is recommended to use the rRNA depletion kit (Cat#4992363/4992364/4992391) to remove rRNA first.
2. Can FFPE samples be used to construct library with this kit?
The mRNA in FFPE samples will be degraded to a certain extent, with relative poor integrity. When using this kit for the library construction, it is recommended to optimize the fragmentation time (shorten the fragmentation time or not performing fragmentation).
3. Using the size selection step provided in the product manual, what might cause the inserted segment to appear slight deviation?
Size selection shall be carried out in strict accordance with the size selection step in this product manual. If there is deviation, the reason might be that the magnetic beads are not balanced to room temperature or are not fully mixed, the pipette is not accurate or the liquid is remained in the tip. It is recommended to use the tips with low adsorption for the experiment.
4. Selection of adapters in library construction
The library construction kit does not contain adapter reagent, and it is recommended to use this kit together with TIANSeq Single-Index Adapter (Illumina)(4992641/4992642/4992378).
5. QC of the library
Library quantitative detection: Qubit and qPCR are used to determine the mass concentration and molar concentration of the library respectively. The operation is strictly in accordance with the product manual. The concentration of the library will generally meet the requirements of NGS sequencing. Detection of library distribution range: Using Agilent 2100 Bioanalyzer to detect the library distribution range.
6. Selection of amplification cycle number
According to the instructions, the number of PCR cycles is 6-12, and the number of PCR cycles needed should be selected according to the sample input. In high-yield libraries, over amplification usually occurs in varying degrees, which is manifested by a slightly larger peak after the peak of the target range in the detection of Agilent 2100 Bioanalyzer, or the detected concentration of Qubit is lower than that of qPCR. Mild over amplification is a normal phenomenon, which does not affect library sequencing and subsequent data analysis.
7. Spikes appears in the detection profile of Agilent 2100 Bioanalyzer
The appearance of spikes in Agilent 2100 Bioanalyzer detection is because of the uneven fragmentation of samples, where there will be more fragments in certain size, and this will become more obvious after PCR enrichment. In this case, it is suggested not to perform the size selection, i.e. set the fragmentation condition to 94°C for 15 min incubated, where the fragment distribution is small and concentrated, and the homogeneity can be improved.







