1. What type of sample is applicable to this kit?
The applicable sample type of this kit can be total RNA or purified mRNA with good RNA integrity. If total RNA is used to construct the library, it is recommended to use the rRNA depletion kit (Cat#4992363/4992364/4992391) to remove rRNA first.
2. Can FFPE samples be used to construct library with this kit?
The mRNA in FFPE samples will be degraded to a certain extent, with relative poor integrity. When using this kit for the library construction, it is recommended to optimize the fragmentation time (shorten the fragmentation time or not performing fragmentation).
3. Using the size selection step provided in the product manual, what might cause the inserted segment to appear slight deviation?
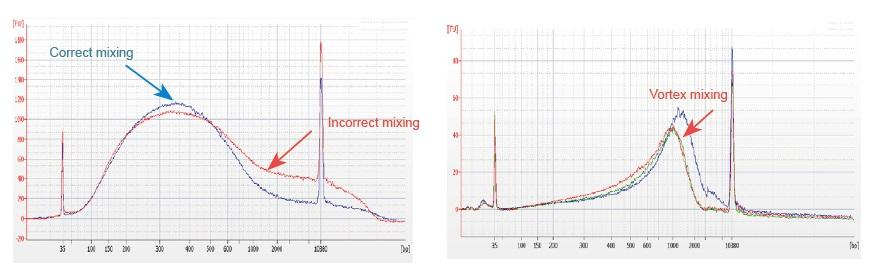
Size selection shall be carried out in strict accordance with the size selection step in this product manual. If there is deviation, the reason might be that the magnetic beads are not balanced to room temperature or are not fully mixed, the pipette is not accurate or the liquid is remained in the tip. It is recommended to use the tips with low adsorption for the experiment.
4. Selection of adapters in library construction
The library construction kit does not contain adapter reagent, and it is recommended to use this kit together with TIANSeq Single-Index Adapter (Illumina)(4992641/4992642/4992378).
5. QC of the library
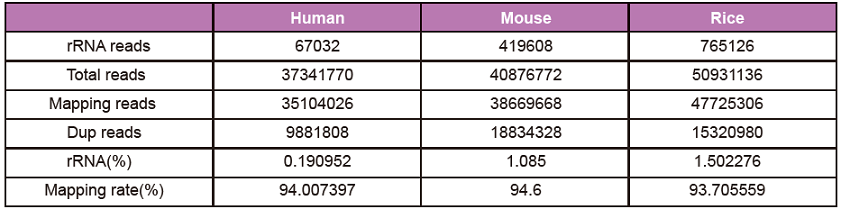
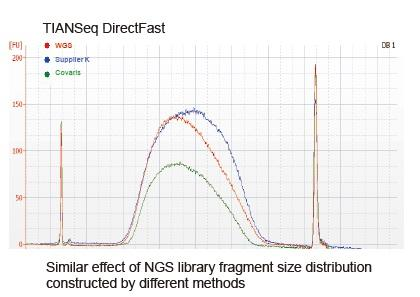
Library quantitative detection: Qubit and qPCR are used to determine the mass concentration and molar concentration of the library respectively. The operation is strictly in accordance with the product manual. The concentration of the library will generally meet the requirements of NGS sequencing. Detection of library distribution range: Using Agilent 2100 Bioanalyzer to detect the library distribution range.
6. Selection of amplification cycle number
According to the instructions, the number of PCR cycles is 6-12, and the number of PCR cycles needed should be selected according to the sample input. In high-yield libraries, over amplification usually occurs in varying degrees, which is manifested by a slightly larger peak after the peak of the target range in the detection of Agilent 2100 Bioanalyzer, or the detected concentration of Qubit is lower than that of qPCR. Mild over amplification is a normal phenomenon, which does not affect library sequencing and subsequent data analysis.
7. Spikes appears in the detection profile of Agilent 2100 Bioanalyzer
The appearance of spikes in Agilent 2100 Bioanalyzer detection is because of the uneven fragmentation of samples, where there will be more fragments in certain size, and this will become more obvious after PCR enrichment. In this case, it is suggested not to perform the size selection, i.e. set the fragmentation condition to 94°C for 15 min incubated, where the fragment distribution is small and concentrated, and the homogeneity can be improved.